
VANFLYTA 26,5 mg, comprimé pelliculé, boîte de 56 plaquettes prédécoupées de 1
Dernière révision : 16/12/2024
Taux de TVA : 2.1%
Prix de vente : 10 700,23 €
Taux remboursement SS : 100%
Base remboursement SS : 10 700,23 €
Laboratoire exploitant : DAIICHI SANKYO FRANCE
Source :

VANFLYTA est indiqué en association avec une chimiothérapie d'induction standard à base de cytarabine et d'anthracycline et avec une chimiothérapie de consolidation standard à base de cytarabine, suivie d'un traitement d'entretien à base de VANFLYTA en monothérapie chez les patients adultes atteints de leucémie aiguë myéloïde (LAM) nouvellement diagnostiquée avec mutation du gène FLT3-ITD.
- Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
- Syndrome du QT long congénital (voir rubrique Mises en garde spéciales et précautions d'emploi).
- Allaitement (voir rubrique Fertilité, grossesse et allaitement).
Allongement de l'intervalle QT
Le quizartinib entraîne un allongement de l'intervalle QT (voir rubrique Effets indésirables). L'allongement de l'intervalle QT peut majorer le risque d'arythmies ventriculaires ou de torsades de pointes. Les patients présentant un syndrome du QT long congénital et/ou ayant des antécédents de torsades de pointes étaient exclus du programme de développement clinique du quizartinib. VANFLYTA ne doit pas être utilisé chez les patients présentant un syndrome du QT long congénital.
VANFLYTA doit être utilisé avec précaution chez les patients ayant un risque significatif d'allongement de l'intervalle QT. Cela comprend les patients présentant une maladie cardiovasculaire sévère ou non contrôlée (par exemple antécédents de bloc cardiaque du deuxième ou troisième degré (sans stimulateur cardiaque/pacemaker), d'infarctus du myocarde au cours des 6 mois précédents, angor non contrôlé, hypertension non contrôlée, insuffisance cardiaque congestive, antécédents d'arythmies ventriculaires ou de torsades de pointes cliniquement significatives) et les patients recevant un traitement concomitant par des médicaments connus pour entraîner un allongement de l'intervalle QT. Les taux d'électrolytes doivent être maintenus dans les valeurs normales (voir rubrique Posologie et mode d'administration).
Le traitement par VANFLYTA ne doit pas être instauré si l'intervalle QTcF est supérieur à 450 ms.
Pendant les phases d'induction et de consolidation, des ECG doivent être réalisés avant l'instauration du traitement, puis une fois par semaine pendant le traitement par le quizartinib ou plus fréquemment si le tableau clinique le justifie.
Pendant la phase d'entretien, des ECG doivent être réalisés avant l'instauration du traitement, puis une fois par semaine pendant le premier mois suivant le début du traitement et une augmentation de la dose, et en fonction du tableau clinique ensuite. La dose d'entretien initiale ne doit pas être augmentée si l'intervalle QTcF est supérieur à 450 ms (voir tableau 1).
Le traitement par VANFLYTA doit être arrêté définitivement chez les patients qui développent un allongement de l'intervalle QT avec des signes ou symptômes d'arythmies engageant le pronostic vital (voir rubrique Posologie et mode d'administration).
Le contrôle ECG de l'intervalle QT doit être réalisé plus fréquemment chez les patients présentant un risque significatif d'allongement de l'intervalle QT et de torsades de pointes.
Les taux de potassium et de magnésium doivent être contrôlés et l'hypokaliémie et l'hypomagnésémie doivent être corrigées avant et pendant le traitement par VANFLYTA. Les valeurs de l'ionogramme et les paramètres ECG doivent être contrôlés plus fréquemment chez les patients présentant une diarrhée ou des vomissements.
Contrôles ECG chez les patients recevant des médicaments entraînant un allongement de l'intervalle QT
Les
contrôles ECG doivent être effectués plus fréquemment en cas
d'administration concomitante de VANFLYTA avec des médicaments connus
pour entraîner un allongement de l'intervalle QT (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Administration concomitante avec des inhibiteurs puissants du CYP3A
La
dose de VANFLYTA doit être réduite en cas d'administration concomitante
avec des inhibiteurs puissants du CYP3A car ceux-ci peuvent augmenter
l'exposition au quizartinib (voir rubriques Posologie et mode d'administration et Interactions avec d'autres médicaments et autres formes d'interactions).
Infections chez les patients âgés
Des infections d'issue fatale sont survenues plus fréquemment lors du traitement par le quizartinib chez les patients âgés (de plus de 65 ans) que chez les patients plus jeunes, en particulier en début de traitement. Les patients âgés de plus de 65 ans doivent être étroitement surveillés afin que l'apparition d'infections sévères pendant la phase d'induction puisse être détectée.
Femmes en âge de procréer/Contraception chez les hommes et les femmes
Selon les données chez l'animal, le quizartinib peut avoir des effets délétères sur l'embryon et le fœtus lorsqu'il est administré pendant la grossesse. Chez les femmes en âge de procréer, un test de grossesse doit être réalisé dans les 7 jours précédant l'instauration du traitement par VANFLYTA. Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par VANFLYTA et pendant au moins 7 mois après l'arrêt du traitement. Les hommes ayant une partenaire en âge de procréer doivent utiliser une contraception efficace pendant le traitement par VANFLYTA et pendant au moins 4 mois après la dernière dose (voir rubrique Fertilité, grossesse et allaitement).
Carte patient
Le prescripteur doit parler des risques du traitement par VANFLYTA avec le/la patient(e). Les patients recevront la carte patient lors de chaque délivrance du médicament (carte incluse dans la boîte du médicament).
Résumé du profil de sécurité
Les effets indésirables les plus fréquents étaient : alanine aminotransférase augmentée (58,9 %), thrombopénie (40,0 %), anémie (37,4 %), diarrhée (37,0 %), nausées (34,0 %), douleur abdominale (29,4 %), céphalée (27,5 %), vomissements (24,5 %) et neutropénie (21,9 %).
Les effets indésirables de grade 3 ou 4 les plus fréquents étaient : thrombopénie (40 %), anémie (35,5 %), neutropénie (21,5 %), alanine aminotransférase augmentée (12,1 %), bactériémie (7,2 %) et infections fongiques (5,7 %). Les effets indésirables graves les plus fréquents dans le bras VANFLYTA étaient : neutropénie (3,0 %), infections fongiques (2,3 %) et infections herpétiques (2,3 %). Les effets indésirables d'issue fatale ont été : infections fongiques (0,8 %) et arrêt cardiaque (0,4 %).
Les effets indésirables les plus fréquents ayant entraîné une interruption du traitement par VANFLYTA étaient : neutropénie (10,6 %), thrombopénie (4,5 %) et intervalle QT allongé à l'électrocardiogramme (2,6 %). Les effets indésirables les plus fréquents ayant entraîné une réduction de la dose étaient : neutropénie (9,1 %), thrombopénie (4,5 %) et intervalle QT allongé à l'électrocardiogramme (3,8 %).
L'effet indésirable le plus fréquent ayant entraîné l'arrêt définitif du traitement par VANFLYTA était la thrombopénie (1,1 %).
Tableau récapitulatif des effets indésirables
La sécurité de VANFLYTA a été évaluée dans l'étude QuANTUM-First, une étude randomisée en double aveugle, contrôlée contre placebo, menée chez des patients adultes atteints de LAM avec mutation FLT3-ITD non préalablement traitée.
Les effets indésirables sont présentés par classe de systèmes d'organes (SOC) MedDRA. Au sein de chaque SOC, les effets indésirables sont classés par ordre décroissant de fréquence selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 4 : Effets indésirables
| Effet indésirable | Tous grades % | Grade 3 ou 4 % | Catégorie de fréquence (tous grades) |
| Infections et infestations | |||
| Infections des voies aériennes supérieuresa | 18,1 | 1,9 | Très fréquent |
| Infections fongiquesb | 15,1 | 5,7 | Très fréquent |
| Infections herpétiquesc | 14,0 | 3,0 | Très fréquent |
| Bactériémied | 11,3 | 7,2 | Très fréquent |
| Affections hématologiques et du système lymphatique | |||
| Thrombopéniee | 40,0 | 40,0 | Très fréquent |
| Anémiee | 37,4 | 35,5 | Très fréquent |
| Neutropéniee | 21,9 | 21,5 | Très fréquent |
| Pancytopénie | 2,6 | 2,3 | Fréquent |
| Troubles du métabolisme et de la nutrition | |||
| Appétit diminué | 17,4 | 4,9 | Très fréquent |
| Affections du système nerveux | |||
| Céphaléef | 27,5 | 0 | Très fréquent |
| Affections cardiaques | |||
| Arrêt cardiaqueg | 0,8 | 0,4 | Peu fréquent |
| Fibrillation ventriculaireg | 0,4 | 0,4 | Peu fréquent |
| Affections respiratoires, thoraciques et médiastinales |
|||
| Épistaxis | 15,1 | 1,1 | Très fréquent |
| Affections gastro-intestinales | |||
| Diarrhéeh | 37,0 | 3,8 | Très fréquent |
| Nausées | 34,0 | 1,5 | Très fréquent |
| Douleur abdominalei | 29,4 | 2,3 | Très fréquent |
| Vomissements | 24,5 | 0 | Très fréquent |
| Dyspepsie | 11,3 | 0,4 | Très fréquent |
| Affections hépatobiliaires | |||
| ALAT augmentéee | 58,9 | 12,1 | Très fréquent |
| Troubles généraux et anomalies au site d'administration |
|||
| Œdèmej | 18,9 | 0,4 | Très fréquent |
| Investigations |
|||
| Intervalle QT allongé à l'électrocardiogrammek |
14,0 | 3,0 | Très fréquent |
a
Le terme « infection des voies aériennes supérieures » inclut :
infection des voies aériennes supérieures, rhinopharyngite, sinusite,
rhinite, angine, laryngopharyngite, pharyngite bactérienne,
pharyngoamygdalite, pharyngite virale et sinusite aiguë.
b Le terme « infections fongiques » inclut : candidose orale, aspergillose bronchopulmonaire, infection fongique, candidose vulvovaginale, infection à Aspergillus, infection fongique des voies aériennes inférieures, mycose orale, infection à Candida, infection cutanée fongique, mucormycose, candidose oropharyngée, aspergillose orale, infection hépatique fongique, candidose hépatosplénique, onychomycose, fongémie, candidose systémique et mycose systémique.
c Le terme « infections herpétiques » inclut : herpès buccal, zona, infections à herpèsvirus, herpès, infection à herpèsvirus humain 6, herpès génital et dermatite herpétique.
d Le terme « bactériémie » inclut : bactériémie, bactériémie à Klebsiella, bactériémie à staphylocoque, bactériémie à entérocoque, bactériémie à streptocoque, bactériémie liée au dispositif, bactériémie à Escherichia, bactériémie à Corynebacterium et bactériémie à Pseudomonas.
e Termes basés sur les données de laboratoire.
f Le terme « céphalée » inclut : céphalée, céphalée de tension et migraine.
g Un patient a présenté deux événements (fibrillation ventriculaire et arrêt cardiaque).
h Le terme « diarrhée » inclut : diarrhée et diarrhée hémorragique.
i Le terme « douleur abdominale » inclut : douleur abdominale, douleur abdominale haute, gêne abdominale, douleur abdominale basse et douleur gastro-intestinale.
j Le terme « œdème » inclut : œdèmes périphériques, œdème de la face, œdème, surcharge liquidienne, œdème généralisé, gonflement périphérique, œdème localisé et gonflement du visage.
k Le terme « intervalle QT allongé à l'électrocardiogramme » inclut : intervalle QT allongé à l'électrocardiogramme et intervalle QT anormal sur l'électrocardiogramme
b Le terme « infections fongiques » inclut : candidose orale, aspergillose bronchopulmonaire, infection fongique, candidose vulvovaginale, infection à Aspergillus, infection fongique des voies aériennes inférieures, mycose orale, infection à Candida, infection cutanée fongique, mucormycose, candidose oropharyngée, aspergillose orale, infection hépatique fongique, candidose hépatosplénique, onychomycose, fongémie, candidose systémique et mycose systémique.
c Le terme « infections herpétiques » inclut : herpès buccal, zona, infections à herpèsvirus, herpès, infection à herpèsvirus humain 6, herpès génital et dermatite herpétique.
d Le terme « bactériémie » inclut : bactériémie, bactériémie à Klebsiella, bactériémie à staphylocoque, bactériémie à entérocoque, bactériémie à streptocoque, bactériémie liée au dispositif, bactériémie à Escherichia, bactériémie à Corynebacterium et bactériémie à Pseudomonas.
e Termes basés sur les données de laboratoire.
f Le terme « céphalée » inclut : céphalée, céphalée de tension et migraine.
g Un patient a présenté deux événements (fibrillation ventriculaire et arrêt cardiaque).
h Le terme « diarrhée » inclut : diarrhée et diarrhée hémorragique.
i Le terme « douleur abdominale » inclut : douleur abdominale, douleur abdominale haute, gêne abdominale, douleur abdominale basse et douleur gastro-intestinale.
j Le terme « œdème » inclut : œdèmes périphériques, œdème de la face, œdème, surcharge liquidienne, œdème généralisé, gonflement périphérique, œdème localisé et gonflement du visage.
k Le terme « intervalle QT allongé à l'électrocardiogramme » inclut : intervalle QT allongé à l'électrocardiogramme et intervalle QT anormal sur l'électrocardiogramme
Description de certains effets indésirables
Affections cardiaques
Le
quizartinib entraîne un allongement de l'intervalle QT sur l'ECG. Des
effets indésirables apparus sous traitement d'allongement de
l'intervalle QT de tout grade ont été rapportés chez 14,0 % des
patients traités par VANFLYTA et 3,0 % des patients ont présenté des
effets indésirables de grade ≥ 3. L'allongement de l'intervalle QT a
entraîné une réduction de la dose chez 10 patients (3,8 %), une
interruption du traitement chez 7 patients (2,6 %) et l'arrêt du
traitement chez 2 patients (0,8 %). Selon une revue centralisée des
données ECG, un allongement de l'intervalle QTcF > 500 ms est
survenu chez 2,3 % des patients. Deux patients (0,8 %) traités par
VANFLYTA ont présenté un arrêt cardiaque avec fibrillation
ventriculaire documentée, d'issue fatale chez un patient, les deux cas
étant survenus dans le contexte d'une hypokaliémie sévère. Des
électrocardiogrammes doivent être réalisés, les taux de
potassium et de magnésium doivent être contrôlés et l'hypokaliémie et
l'hypomagnésémie doivent être corrigées avant et pendant le traitement
par VANFLYTA. Pour les modifications de la posologie chez les patients
présentant un allongement de l'intervalle QT, voir la rubrique Posologie et mode d'administration.
Autres populations particulières
Sujets âgés
Des
infections d'issue fatale sont survenues plus fréquemment lors du
traitement par le quizartinib chez les patients âgés (de plus de 65
ans) que chez les patients plus jeunes (13 % versus 5,7 %), en particulier en début de traitement.
Les patients âgés de plus de 65 ans doivent être étroitement surveillés afin que l'apparition d'infections sévères pendant la phase d'induction puisse être détectée.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT INSTAURATION DU TRAITEMENT par quizartinib :
- Confirmer la présence de la mutation FLT3-ITD à l'aide d'un
dispositif médical de diagnostic in vitro (DIV) avec marquage CE pour
l'usage prévu correspondant. Si un tel dispositif de DIV n'est pas
disponible, la positivité pour la mutation FLT3-ITD doit être confirmée
à l'aide d'un autre test validé.
- Réaliser des ECG et corriger les anomalies électrolytiques
(hypokaliémie et l'hypomagnésémie). N'instaurer le traitement que si
l'intervalle QTcF est inférieur ou égal à 450 ms.
- Réaliser un test de grossesse dans les 7 jours précédant l'instauration du traitement chez la femme en âge de procréer.
SURVEILLANCE :
- Réaliser des ECG une fois par semaine pendant les phases d'induction
et de consolidation par le quizartinib, ou plus fréquemment si le
tableau clinique le justifie.
- Réaliser des ECG une fois par semaine pendant le premier mois suivant
le début de la phase d'entretien et une augmentation de la dose, et en
fonction du tableau clinique ensuite.
- Corriger les anomalies électrolytiques (hypokaliémie et l'hypomagnésémie).
Chez les patients devant recevoir une greffe de
cellules souches hématopoïétiques (greffe de CSH), ARRETER le traitement par
quizartinib 7 jours avant le début d'un protocole de conditionnement.
INFORMER IMMEDIATEMENT le médecin en cas de :
- Sensations vertigineuses, étourdissements ou impression d'être sur le point de s'évanouir.
- Fèvre, toux, douleur thoracique, essoufflement, fatigue ou douleur en urinant.
Eviter la prise de produits à base de plantes contenant du millepertuis (Hypericum perforatum).
FEMME EN AGE DE PROCREER : utiliser une contraception efficace pendant
le traitement par quizartinib et pendant au moins 7 mois après l'arrêt
du traitement.
HOMME ayant une partenaire en âge de procréer : utiliser une
contraception efficace pendant le traitement par quizartinib et pendant
au moins 4 mois après l'arrêt du traitement.
CONSERVER la parte patient en permanence sur
soi.
Femmes en âge de procréer/Contraception chez les hommes et les femmes
Chez les femmes en âge de procréer, un test de grossesse doit être réalisé dans les 7 jours précédant l'instauration du traitement par VANFLYTA.
Le quizartinib peut avoir des effets délétères sur l'embryon et le fœtus lorsqu'il est administré pendant la grossesse (voir rubrique Données de sécurité préclinique) ; par conséquent, les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par VANFLYTA et pendant au moins 7 mois après la dernière dose.
Les hommes ayant une partenaire en âge de procréer doivent utiliser une contraception efficace pendant le traitement par VANFLYTA et pendant au moins 4 mois après la dernière dose.
Grossesse
Il n'existe pas de données sur l'utilisation du quizartinib chez la femme enceinte. Selon les données chez l'animal, le quizartinib peut provoquer une toxicité embryonnaire et fœtale lorsqu'il est administré pendant la grossesse (voir rubrique Données de sécurité préclinique).
VANFLYTA ne doit pas être utilisé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas une contraception efficace, à moins que la situation clinique de la femme ne justifie le traitement. Les femmes enceintes doivent être informées du risque potentiel pour le fœtus.
Allaitement
On ne sait pas si le quizartinib ou ses métabolites actifs sont excrétés dans le lait maternel. Un risque pour les nouveau-nés/nourrissons ne peut être exclu. Compte tenu du risque d'effets indésirables graves chez les enfants allaités, les femmes ne doivent pas allaiter pendant le traitement par VANFLYTA et pendant au moins cinq semaines après la dernière dose (voir rubrique Contre-indications).
Fertilité
Il n'existe pas de données concernant l'effet du quizartinib sur la fertilité humaine. Selon les données chez l'animal, le traitement par VANFLYTA peut altérer la fertilité chez les hommes et les femmes (voir rubrique Données de sécurité préclinique).
In vitro, le quizartinib et son métabolite actif AC886 sont métabolisés principalement par le CYP3A.
Effet d'autres médicaments sur VANFLYTA
Inhibiteurs puissants du CYP3A et de la glycoprotéine P (P-gp)
L'administration concomitante de kétoconazole, un inhibiteur puissant du CYP3A et de la P-gp, (200 mg deux fois par jour pendant 28 jours) avec une dose unique de VANFLYTA a entraîné une augmentation de 1,17 fois et 1,94 fois respectivement de la concentration plasmatique maximale (Cmax) et de l'aire sous la courbe (ASCinf) du quizartinib et une diminution de 2,5 fois et 1,18 fois respectivement de la Cmax et de l'ASCinf de l'AC886, par rapport aux valeurs observées après administration de VANFLYTA seul. Il a été estimé qu'à l'état d'équilibre, l'exposition au quizartinib (Cmax et ASC0-24 h) était augmentée de 1,86 fois et 1,96 fois respectivement et que l'exposition à l'AC886 (Cmax et ASC0-24 h) était diminuée de 1,22 fois et 1,17 fois respectivement. L'augmentation de l'exposition au quizartinib peut majorer le risque de toxicité.
Si l'administration concomitante d'inhibiteurs puissants du CYP3A ne peut être évitée, la dose de VANFLYTA doit être réduite comme il est indiqué dans le tableau ci-dessous. Pour des informations supplémentaires concernant les ajustements de la posologie, voir le tableau 3 à la rubrique Posologie et mode d'administration.
| Pleine dose | Réductions de dose en cas d'administration concomitante d'inhibiteurs puissants du CYP3A |
| 26,5 mg | 17,7 mg |
| 35,4 mg | |
| 53 mg | 26,5 mg |
Les inhibiteurs puissants du CYP3A et de la P-gp sont par exemple l'itraconazole, le posaconazole, le voriconazole, la clarithromycine, la néfazodone, la télithromycine et les antirétroviraux (certains médicaments utilisés pour traiter le VIH peuvent soit augmenter le risque d'effets indésirables [par exemple, le ritonavir], soit réduire l'efficacité [par exemple, l'éfavirenz ou l'étravirine] de VANFLYTA).
Inhibiteurs modérés du CYP3A
L'administration
concomitante de fluconazole, un inhibiteur modéré du CYP3A, (200 mg
deux fois par jour pendant 28 jours) avec une dose unique de VANFLYTA a
entraîné une augmentation de 1,11 fois et 1,02 fois respectivement de la Cmax et de 1,20 fois et 1,14 fois respectivement de l'ASCinf du
quizartinib et de l'AC886. Ces modifications n'ont pas été considérées
comme cliniquement pertinentes. Aucune modification de la posologie
n'est recommandée.
Inducteurs puissants ou modérés du CYP3A
L'administration
concomitante d'éfavirenz, un inducteur modéré du CYP3A, (phase
d'instauration du traitement à la dose de 600 mg une fois par jour
pendant 14 jours) avec une dose unique de VANFLYTA a entraîné une diminution d'environ 1,18 fois et 9,7 fois respectivement de la Cmax et de l'ASCinf du quizartinib par rapport aux valeurs observées après administration de VANFLYTA seul. La Cmax et l'ASCinf de l'AC886 étaient diminuées d'environ 3,1 fois et 26 fois respectivement (voir rubrique Propriétés pharmacocinétiques).
La diminution de l'exposition au quizartinib peut entraîner une diminution de l'efficacité. L'administration concomitante de VANFLYTA avec des inducteurs puissants ou modérés du CYP3A doit être évitée.
Les inducteurs puissants du CYP3A4 sont par exemple l'apalutamide, la carbamazépine, l'enzalutamide, le mitotane, la phénytoïne, la rifampicine et certains médicaments à base de plantes tels que le millepertuis (Hypericum perforatum). Les inducteurs modérés du CYP3A4 sont par exemple l'éfavirenz, le bosentan, l'étravirine, le phénobarbital et la primidone.
Médicaments entraînant un allongement de l'intervalle QT
L'administration
concomitante de VANFLYTA avec d'autres médicaments qui entraînent un
allongement de l'intervalle QT peut augmenter l'incidence d'allongement
de l'intervalle QT. Les médicaments entraînant un allongement de
l'intervalle QT sont par exemple, mais sans s'y limiter, les
antifongiques azolés, l'ondansétron, le granisétron, l'azithromycine,
la pentamidine, la doxycycline, la moxifloxacine, l'atovaquone, la
prochlorpérazine et le tacrolimus. La prudence s'impose en cas
d'administration concomitante de médicaments entraînant un allongement
de l'intervalle QT avec VANFLYTA (voir rubrique Mises en garde spéciales et précautions d'emploi).
Agents réducteurs de l'acidité gastrique
Le lansoprazole, un inhibiteur de la pompe à protons, a entraîné une diminution de 1,16 fois et 1,05 fois respectivement de la Cmax et de l'ASCinf du quizartinib. Cette diminution de l'absorption du quizartinib n'a pas été considérée comme cliniquement pertinente. Aucune modification de la posologie n'est recommandée.
Effet de VANFLYTA sur d'autres médicaments
Substrats de la glycoprotéine P (P-gp)
L'administration concomitante de quizartinib et de dabigatran étexilate (un substrat de la P-gp) a entraîné une augmentation de 1,12 fois et 1,13 fois respectivement de la Cmax et de 1,13 fois et 1,11 fois respectivement de l'ASCinf du dabigatran total et libre (voir rubrique Propriétés pharmacocinétiques). Le quizartinib est un
inhibiteur faible de la P-gp et aucune modification de la posologie
n'est recommandée en cas d'administration concomitante de substrats de
la P-gp avec VANFLYTA.
Substrats de la protéine de résistance du cancer du sein (breast cancer resistance protein, BCRP)
Des données in vitro indiquent
que le quizartinib est un inhibiteur de la BCRP. La pertinence clinique
n'est pas connue actuellement. La prudence s'impose en cas
d'administration concomitante de quizartinib avec des médicaments qui
sont des substrats de la BCRP.
Le traitement par VANFLYTA doit être instauré par un médecin expérimenté dans l'utilisation des traitements anticancéreux.
Avant
le début du traitement par VANFLYTA chez les patients atteints de LAM,
la présence de la mutation FLT3-ITD doit être confirmée à l'aide d'un
dispositif médical de diagnostic in vitro (DIV) avec marquage
CE pour l'usage prévu correspondant. Si un tel dispositif de DIV n'est
pas disponible, la positivité pour la mutation FLT3-ITD doit être
confirmée à l'aide d'un autre test validé.
Avant
le début du traitement, des ECGs doivent être réalisés et les anomalies
électrolytiques doivent être corrigées (voir rubrique Mises en garde spéciales et précautions d'emploi).
Posologie
VANFLYTA doit être administré en association avec la chimiothérapie standard à la dose de 35,4 mg (2 × 17,7 mg) une fois par jour pendant deux semaines lors de chaque cycle d'induction. Chez les patients qui obtiennent une rémission complète (RC) ou une rémission complète avec récupération hématologique incomplète (RCi), VANFLYTA doit être administré à la dose de 35,4 mg une fois par jour pendant deux semaines lors de chaque cycle de chimiothérapie de consolidation, puis en monothérapie en traitement d'entretien à une dose initiale de 26,5 mg une fois par jour. Après deux semaines, la dose d'entretien doit être augmentée à 53 mg (2 × 26,5 mg) une fois par jour si l'intervalle QT corrigé selon la formule de Fridericia (QTcF) est ≤ 450 ms (voir tableau 2 et rubrique Mises en garde spéciales et précautions d'emploi). Le traitement d'entretien en monothérapie peut être poursuivi pendant une durée allant jusqu'à 36 cycles.
Pour des informations supplémentaires sur la posologie, voir les tableaux 1 à 3.
Tableau 1 : Schéma posologique
| Instauration du traitement par VANFLYTA | Inductiona | Consolidationb | Entretien |
| À partir du jour 8 (pour le schéma 7 + 3)c | À partir du jour 6 | Premier jour de traitement d'entretien | |
| Dose | 35,4 mg une fois par jour | 35,4 mg une fois par jour |
|
| Durée de traitement (cycles de 28 jours) | Deux semaines lors de chaque cycle | Deux semaines lors de chaque cycle | Une fois par jour sans pause entre les cycles pendant une durée allant jusqu'à 36 cycles. |
a Les patients peuvent recevoir jusqu'à deux cycles de chimiothérapie d'induction.
b Les patients peuvent recevoir jusqu'à quatre cycles de chimiothérapie de consolidation.
c Pour le schéma 5 + 2 en second cycle de chimiothérapie d'induction, le traitement par VANFLYTA doit être débuté le jour 6.
b Les patients peuvent recevoir jusqu'à quatre cycles de chimiothérapie de consolidation.
c Pour le schéma 5 + 2 en second cycle de chimiothérapie d'induction, le traitement par VANFLYTA doit être débuté le jour 6.
Greffe de cellules souches hématopoïétiques
Chez
les patients devant recevoir une greffe de cellules souches
hématopoïétiques (greffe de CSH), le traitement par VANFLYTA doit être
arrêté 7 jours avant le début d'un protocole de conditionnement. Il
peut être repris après la greffe, conformément aux recommandations
posologiques ci-dessus, en fonction du taux de leucocytes et à
l'appréciation du médecin traitant, chez les patients qui présentent
une récupération hématologique suffisante et une réaction du greffon
contre l'hôte (GvH) de grade ≤ 2 ne nécessitant pas l'instauration d'un
nouveau traitement systémique de la GvH dans les 21 jours.
Modifications posologiques
Le traitement par VANFLYTA ne doit être instauré que si l'intervalle QTcF est ≤ 450 ms (voir rubrique Mises en garde spéciales et précautions d'emploi).
Pour les modifications posologiques recommandées en cas d'effets indésirables, voir le tableau 2. Pour les ajustements de la posologie en cas d'effets indésirables et/ou d'administration concomitante d'inhibiteurs puissants du CYP3A, voir le tableau 3.
Tableau 2 : Modifications posologiques recommandées en cas d'effets indésirables
| Effet indésirable | Action recommandée |
| Intervalle QTcF de 450 à 480 ms (grade 1) |
|
| Intervalle QTcF de 481 à 500 ms (grade 2) |
|
| Intervalle QTcF ≥ 501 ms (grade 3) |
|
| Intervalle QTcF ≥ 501 ms récurrent (grade 3) |
|
| Torsades de pointes, tachycardie ventriculaire polymorphe, signes/symptômes d'arythmies engageant le pronostic vital (grade 4) |
|
| Effets indésirables non hématologiques de grade 3 ou 4 |
|
| Neutropénie ou thrombopénie de grade 4 persistante sans atteinte médullaire |
|
Les
grades sont évalués selon les Critères de terminologie communs pour les
événements indésirables du National Cancer Institute version 4.03 (NCI
CTCAE v4.03).
Ajustements de la posologie en cas d'effets indésirables et/ou d'administration concomitante d'inhibiteurs puissants du CYP3A
Tableau 3 : Ajustements de la posologie par phase de traitement en cas d'effets indésirables et/ou d'administration concomitante d'inhibiteurs puissants du CYP3A pendant le traitement par VANFLYTA
| Phase de traitement | Pleine dose | Réductions de dose | ||
| Effet indésirable | Administration concomitante d'inhibiteurs puissants du CYP3A | Effet indésirable et administration concomitante d'inhibiteurs puissants du CYP3A | ||
| Induction ou consolidation | 35,4 mg | 26,5 mg | 17,7 mg | Interrompre le traitement |
| Entretien (deux premières semaines) | 26,5 mg | Interrompre le traitement | 17,7 mg | Interrompre le traitement |
| Entretien (après deux semaines) | 53 mg | 35,4 mg | 26,5 mg | 17,7 mg |
Oubli d'une dose ou vomissements
Si
une dose de VANFLYTA a été oubliée ou n'a pas été prise à l'heure
habituelle, le patient doit la prendre le plus tôt possible le même
jour, puis prendre la dose suivante au moment habituel le lendemain. Le
patient ne doit pas prendre deux doses le même jour.
Si le patient vomit après avoir pris VANFLYTA, il ne doit pas prendre une autre dose ce jour-là, mais prendre la dose suivante le lendemain au moment habituel.
Populations particulières
Sujets âgés
Aucun ajustement de la posologie n'est nécessaire chez les patients âgés.
Insuffisance hépatique
Aucun ajustement de la posologie n'est recommandé chez les patients présentant une insuffisance hépatique légère ou modérée.
VANFLYTA n'est pas recommandé chez les patients atteints d'insuffisance hépatique sévère (classe C de Child-Pugh), car la sécurité et l'efficacité n'ont pas été établies dans cette population.
Insuffisance rénale
Aucun ajustement de la posologie n'est recommandé chez les patients présentant une insuffisance rénale légère ou modérée.
VANFLYTA n'est pas recommandé chez les patients atteints d'insuffisance rénale sévère (ClCr < 30 mL/min estimée selon la formule de Cockcroft-Gault), car la sécurité et l'efficacité n'ont pas été établies dans cette population.
Population pédiatrique
La
sécurité et l'efficacité de VANFLYTA chez les enfants et adolescents
âgés de moins de 18 ans n'ont pas été établies (voir rubrique Propriétés pharmacodynamiques). Aucune donnée n'est disponible.
Mode d'administration
Voie orale.
Les comprimés doivent être pris à peu près à la même heure chaque jour, au cours ou en dehors des repas.
Durée de conservation :
5 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Il n'existe pas d'antidote connu pour un surdosage de VANFLYTA. En cas de surdosage important, des mesures de soutien doivent être mises en place comme nécessaire, avec interruption du traitement, hémogramme et surveillance ECG, contrôle de l'ionogramme et prise en compte des médicaments concomitants susceptibles de prédisposer les patients à un allongement de l'intervalle QT et/ou à des torsades de pointes. Les patients doivent recevoir un traitement symptomatique et de support (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Classe pharmacothérapeutique : Antinéoplasiques, inhibiteurs de protéines kinases, code ATC : L01EX11
Mécanisme d'action
Le quizartinib est un inhibiteur du récepteur à activité tyrosine kinase FLT3. Le quizartinib et son principal métabolite AC886 se lient compétitivement à la poche de liaison adénosine triphosphate (ATP) du récepteur FLT3 avec une affinité élevée. Le quizartinib et l'AC886 inhibent l'activité kinase du récepteur FLT3 en empêchant l'autophosphorylation du récepteur, ce qui inhibe ensuite la signalisation en aval par le récepteur FLT3 et bloque la prolifération cellulaire dépendante du récepteur muté FLT3-ITD.
Effets pharmacodynamiques
Électrophysiologie cardiaque
Selon
l'analyse exposition-réponse de l'étude QuANTUM-First, il est prédit un
allongement de l'intervalle QTcF dépendant de la concentration de 24,1
ms [limite supérieure de l'intervalle de confiance (IC) à 90 % bilatéral : 26,6 ms] à la Cmax du quizartinib à l'état d'équilibre (dose de 53 mg) pendant le traitement d'entretien.
Efficacité et sécurité cliniques
L'efficacité et la sécurité du quizartinib versus placebo ont été évaluées dans une étude de phase III randomisée en double aveugle, contrôlée contre placebo, l'étude QuANTUM-First. Dans l'étude ont été inclus 539 patients adultes âgés de 18 à 75 ans (25 % étaient âgés de 65 ans et plus) présentant une LAM avec mutation FLT3-ITD non préalablement traitée ; le statut mutationnel FLT3-ITD était déterminé prospectivement à l'aide d'un test spécifique à l'étude clinique. Les patients ont été randomisés (1:1) pour recevoir VANFLYTA 35,4 mg une fois par jour (n = 268) ou le placebo (n = 271) pendant deux semaines lors de chaque cycle, en association avec une chimiothérapie standard (traitement d'induction suivi d'un traitement de consolidation chez les patients répondeurs), puis un traitement d'entretien par VANFLYTA en monothérapie (26,5 mg une fois par jour pendant deux semaines et 53 mg une fois par jour ensuite) ou le placebo pendant une durée allant jusqu'à 36 cycles (cycles de 28 jours).
Les patients recevaient jusqu'à deux cycles de chimiothérapie d'induction consistant en daunorubicine ou idarubicine administrée les jours 1, 2 et 3 et cytarabine administrée pendant 7 jours, suivie après la rémission d'un traitement comprenant jusqu'à 4 cycles de chimiothérapie de consolidation et/ou une greffe de CSH. La chimiothérapie de consolidation consistait en cytarabine administrée les jours 1, 3 et 5. Chez les patients devant recevoir une greffe de CSH, le traitement expérimental était arrêté 7 jours avant le début d'un protocole de conditionnement. Pour les recommandations posologiques concernant la daunorubicine, l'idarubicine et la cytarabine, se reporter au Résumé des Caractéristiques du Produit de ces médicaments.
Les deux groupes de traitement randomisé étaient bien équilibrés en termes de caractéristiques démographiques et cliniques et de facteurs de stratification initiaux. Chez les 539 patients, l'âge médian était de 56 ans (plage : 20 à 75 ans), 26,1 % des patients du bras quizartinib et 24 % des patients du bras placebo étaient âgés de 65 ans et plus, 54,5 % étaient des femmes et 45,5 % étaient des hommes, 59,7 % étaient blancs, 29,3 % étaient asiatiques, 1,3 % étaient noirs ou afro-américains et 9,7 % appartenaient à d'autres groupes ethniques. Quatre-vingt-quatre pour cent des patients avaient un indice de performance ECOG (Eastern Cooperative Oncology Group) de 0 ou 1 lors de l'inclusion. La majorité des patients (72,4 %) avaient un statut cytogénétique de risque intermédiaire lors de l'inclusion. La fréquence allélique de la mutation FLT3-ITD était de 3 à 25 % chez 35,6 % des patients, supérieure à 25-50 % chez 52,1 % des patients et supérieure à 50 % chez 12,1 % des patients.
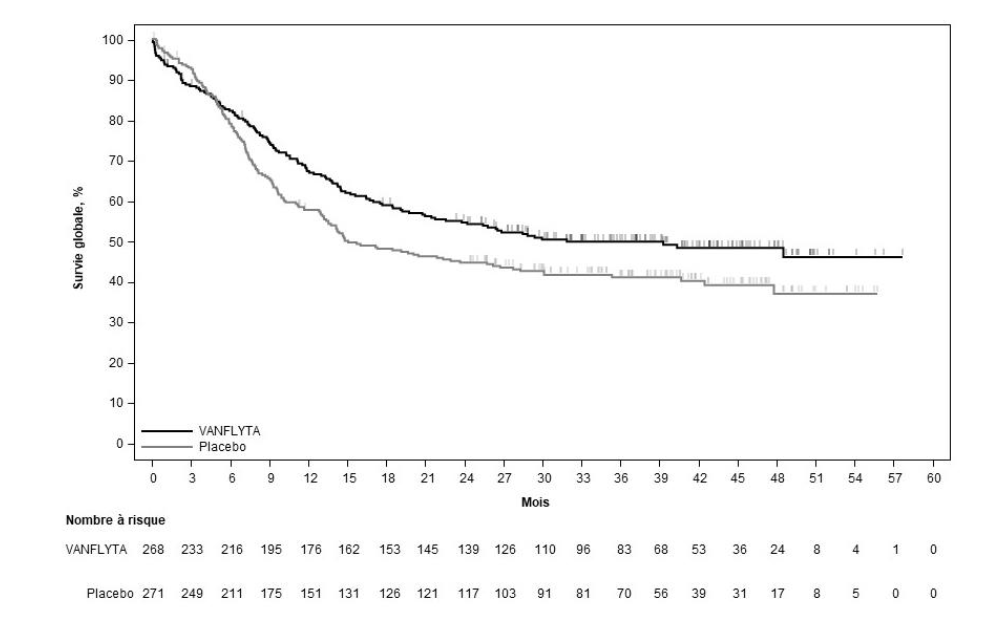
Le critère d'évaluation principal de l'efficacité était la survie globale (SG), définie comme le délai entre la randomisation et le décès de toute cause.
L'étude a montré une amélioration statistiquement significative de la SG dans le bras quizartinib (voir tableau 5 et figure 1). La durée médiane de suivi dans l'étude était de 39,2 mois.
Une différence des estimations des taux de survie (IC à 95 %) entre le bras quizartinib et le bras placebo a été observée aux temps d'évaluation principaux des mois 12, 24, 36 et 48 (voir tableau 5).
Le taux de rémission complète (RC) [IC à 95 %] était de 54,9 % (147/268) [48,7 ; 60,9] avec le quizartinib contre 55,4 % (150/271) [49,2 ; 61,4] avec le placebo.
Tableau 5 : Résultats d'efficacité de l'étude QuANTUM-First (population en intention de traiter)
| Quizartinib N = 268 | Placebo N = 271 | |
| SG (mois) | ||
| Médiane (IC à 95 %)a | 31,9 (21,0 ; NE) | 15,1 (13,2 ; 26,2) |
| HRb par rapport au placebo (IC à 95 %) | 0,776 (0,615 ; 0,979) | |
| Valeur p (test du log-rank stratifié bilatéral) | 0,0324 | |
| Taux de SG (%) (IC à 95 %)a | ||
| Mois 12 | 67,4 (61,3 ; 72,7) | 57,7 (51,6 ; 63,4) |
| Mois 24 | 54,7 (48,4 ; 60,5) | 44,7 (38,7 ; 50,6) |
| Mois 36 | 49,9 (43,7 ; 55,9) | 41,1 (35,0 ; 47,0) |
| Mois 48 | 48,4 (41,9 ; 54,5) | 37,0 (29,8, 44,2) |
IC = intervalle de confiance ; NE = non estimable.
a Estimation selon la méthode de Kaplan-Meier.
b Hazard ratio (HR) déterminé à l'aide d'un modèle de régression de Cox stratifié.
a Estimation selon la méthode de Kaplan-Meier.
b Hazard ratio (HR) déterminé à l'aide d'un modèle de régression de Cox stratifié.
Figure 1 : Courbes de Kaplan-Meier de la survie globale dans l'étude QuANTUM-First
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec VANFLYTA dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement de la leucémie aiguë myéloïde (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
La pharmacocinétique du quizartinib et de son métabolite actif AC886 a été évaluée chez des adultes volontaires sains (dose unique) et chez des patients atteints de LAM non préalablement traitée (à l'état d'équilibre).
Absorption
La biodisponibilité absolue du quizartinib administré sous forme de comprimé était de 71 %. Après administration par voie orale à jeun chez des volontaires sains, le temps jusqu'à la concentration maximale (tmax) médian du quizartinib et de l'AC886 mesuré post-dose était respectivement de 4 heures (plage : 2 à 8 heures) et de 5 à 6 heures (plage : 4 à 120 heures).
Après administration de quizartinib avec des aliments chez des volontaires sains, la Cmax du quizartinib était diminuée de 1,09 fois, l'ASCinf était augmentée de 1,08 fois et le tmax était prolongé de deux heures. Ces modifications de l'exposition ne sont pas considérées comme cliniquement significatives. VANFLYTA peut être administré au cours ou en dehors des repas.
Selon un modèle de pharmacocinétique de population chez des patients atteints de LAM non préalablement traitée, pendant le traitement d'induction à la dose de 35,4 mg par jour, à l'état d'équilibre, les moyennes géométiques (coefficient de variation [%CV]) des Cmax du quizartinib et de l'AC886 estimées étaient respectivement de 140 ng/mL (71 %) et 163 ng/mL (52 %) et les moyennes géométriques (%CV) des ASC0-24 h étaient respectivement de 2 680 ng•h/mL (85 %) et 3 590 ng•h/mL (51 %).
Pendant le traitement de consolidation à la dose de 35,4 mg par jour, à l'état d'équilibre, les moyennes géométiques (%CV) des Cmax du quizartinib et de l'AC886 estimées étaient respectivement de 204 ng/mL (64 %) et 172 ng/mL (47 %) et les moyennes géométriques (%CV) des ASC0-24 h étaient respectivement de 3 930 ng•h/mL (78 %) et 3 800 ng•h/mL (46 %).
Pendant le traitement d'entretien à la dose de 53 mg par jour, à l'état d'équilibre, les moyennes géométriques (coefficient de variation [%CV]) des Cmax du quizartinib et de l'AC886 estimées étaient respectivement de 529 ng/mL (60 %) et 262 ng/mL (48 %) et les moyennes géométriques (%CV) des ASC0-24 h étaient respectivement de 10 200 ng•h/mL (75 %) et 5 790 ng•h/mL (46 %).
Distribution
In vitro, la liaison du quizartinib et de l'AC886 aux protéines plasmatiques humaines est ≥ 99 %.
Les rapports sang/plasma du quizartinib et de l'AC886 dépendent de la concentration, ce qui indique une saturation de la distribution dans les érythrocytes. Aux concentrations plasmatiques cliniquement pertinentes, le rapport sang/plasma est d'environ 1,3 pour le quizartinib et d'environ 2,8 pour l'AC886. Le rapport sang/plasma de l'AC886 dépend également de l'hématocrite, avec une tendance à l'augmentation à des taux d'hématocrite élevés.
Chez les volontaires sains, la moyenne géométrique (%CV) du volume de distribution du quizartinib était estimée à 275 L (17 %).
Biotransformation
In vitro, le quizartinib est métabolisé principalement par le CYP3A4 et le CYP3A5 par des voies oxydatives pour former le métabolite actif AC886, qui est ensuite métabolisé par les mêmes isoenzymes CYP3A4 et CYP3A5. Le rapport des ASC0-24 h AC886/quizartinib à l'état d'équilibre pendant le traitement d'entretien était de 0,57.
Élimination
Les demi-vies (t1/2) effectives moyennes (écart-type [ET]) du quizartinib et de l'AC886 sont respectivement de 81 heures (73) et 136 heures (113) chez les patients atteints de LAM non préalablement traitée. Les rapports d'accumulation moyens (ET) (ASC0-24 h) du quizartinib et de l'AC886 étaient respectivement de 5,4 (4,4) et 8,7 (6,8).
Le quizartinib et ses métabolites sont éliminés principalement par voie hépatobiliaire, avec une excrétion essentiellement dans les fèces (76,3 % de la dose radioactive administrée par voie orale). Dans les fèces, le quizartinib sous forme inchangée représentait environ 4 % de la dose radioactive administrée par voie orale. L'excrétion rénale était une voie d'élimination mineure du quizartinib radiomarqué (moins de 2 % de la dose administrée).
Chez les volontaires sains, la moyenne géométrique (%CV) de la clairance corporelle totale du quizartinib était estimée à 2,23 L/heure (29 %).
Linéarité/non-linéarité
La pharmacocinétique du quizartinib et de l'AC886 est linéaire dans l'intervalle de doses de 26,5 mg à 79,5 mg chez les volontaires sains et de 17,7 mg à 53 mg chez les patients atteints de LAM.
Relations pharmacocinétique/pharmacodynamique
Selon une analyse pharmacocinétique de population, l'âge (18 à 91 ans), le groupe ethnique, le sexe, le poids ou l'insuffisance rénale (ClCr de 30 à 89 mL/min estimée selon la formule de Cockcroft-Gault) n'ont pas d'effet cliniquement significatif sur l'exposition au quizartinib et à l'AC886.
Études d'interactions avec d'autres médicaments
Transporteurs
Les études in vitro ont
montré que le quizartinib est un substrat de la P-gp, mais pas de la
BCRP, d'OATP1B1, d'OATP1B3, d'OCT1, d'OAT2, de MATE1 ni de la MRP2.
L'AC886 est un substrat de la BCRP, mais pas d'OATP1B1, d'OATP1B3, de
MATE1 ni de la MRP2. Cependant l'administration d'une dose unique de
quizartinib avec le kétoconazole, un inhibiteur puissant du CYP3A et de
la P-gp, a entraîné une augmentation d'environ 1,17 fois de la Cmax du quizartinib, ce qui semble indiquer que l'effet
de la P-gp est minime. Un ajustement de la posologie étant nécessaire
en cas d'administration concomitante d'inhibiteurs puissants du CYP3A,
dont de nombreux sont également des inhibiteurs de la P-gp, aucun
ajustement de la posologie spécifique n'est nécessaire en cas
d'utilisation d'inhibiteurs de la P-gp.
Substrats de la protéine de résistance du cancer du sein (breast cancer resistance protein, BCRP)
Le quizartinib est un inhibiteur de la BCRP, avec une CI50 estimée de 0,813 µM in vitro. En l'absence de
données cliniques, il ne peut être exclu qu'aux doses recommandées, le
quizartinib puisse être un inhibiteur de ce transporteur.
Substrats de l'uridine diphosphate glucuronyltransférase (UGT) 1A1
Le quizartinib est un inhibiteur de l'UGT1A1, avec un Ki estimé de 0,78 µM in vitro. Selon l'analyse d'un modèle pharmacocinétique physiologique (PBPK), il est prédit que le quizartinib entraîne une augmentation de 1,03 fois de la Cmax et de l'ASCinf du raltégravir (un substrat de l'UGT1A1), qui n'est pas considérée comme cliniquement pertinente.
Populations particulières
Insuffisance hépatique
Dans
une étude de phase I à dose unique (26,5 mg), la pharmacocinétique du
quizartinib et de l'AC886 a été évaluée chez des patients présentant
une insuffisance hépatique légère (classe A de Child-Pugh) ou modérée
(classe B de Child-Pugh) et comparée à la pharmacocinétique chez des
sujets ayant une fonction hépatique normale. L'exposition (Cmax et ASCinf) au quizartinib et à l'AC886 était similaire
(différence ≤ 30 %) dans tous les groupes. L'insuffisance hépatique n'a
pas d'effet sur la liaison aux protéines du quizartinib et de l'AC886.
Par conséquent, l'insuffisance hépatique n'a pas d'effet cliniquement
significatif sur l'exposition au quizartinib et à l'AC886.
Aucun ajustement de la posologie n'est recommandé chez les patients présentant une insuffisance hépatique légère ou modérée.
Il n'était pas inclus de patients atteints d'insuffisance hépatique sévère (classe C de Child-Pugh) dans les études cliniques ; par conséquent, VANFLYTA n'est pas recommandé chez ces patients.
Insuffisance rénale
Une
analyse pharmacocinétique de population a montré que la fonction rénale
n'a pas d'influence sur la clairance du quizartinib et de l'AC886 chez
les patients atteints de LAM présentant une insuffisance rénale légère
à modérée (ClCr de 30 à 89 mL/min). Par conséquent, l'insuffisance
rénale légère ou modérée n'a pas d'effet cliniquement significatif sur
l'exposition au quizartinib et à l'AC886. Aucun ajustement de la
posologie n'est recommandé chez les patients présentant une
insuffisance rénale légère ou modérée.
Les patients présentant une insuffisance rénale sévère (ClCr < 30 mL/min) étaient exclus des études cliniques ; par conséquent, VANFLYTA n'est pas recommandé chez ces patients.
VANFLYTA n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Dans les études de génotoxicité, le quizartinib a été mutagène dans un essai de mutation réverse sur bactéries, mais pas dans un essai de mutation sur cellules de mammifère (test du lymphome de souris utilisant le gène de la thymidine kinase) ni dans un essai de mutations de cellules de rongeurs transgéniques in vivo. Le quizartinib n'a pas été clastogène et n'a pas induit de polyploïdie dans un essai d'aberration chromosomique et n'a pas été clastogène ni aneugène dans le test des micronoyaux sur cellules de moelle osseuse à dose unique chez le rat. Le résultat d'un test des micronoyaux sur cellules de moelle osseuse in vivo chez le rat a été équivoque après administration répétée pendant 28 jours. Après administration d'une dose unique plus élevée, le résultat a été négatif.
Il n'a pas été réalisé d'études de la fertilité chez l'animal avec le quizartinib. Cependant, des effets nocifs sur les systèmes reproducteurs mâle et femelle ont été observés dans les études de toxicologie en administration répétée effectuées chez le rat et le singe. Chez la rate, des kystes ovariens et des modifications de la muqueuse vaginale ont été constatés aux doses représentant environ 10 fois la dose recommandée chez l'homme (DRH) sur la base de l'ASC. Les anomalies chez le singe femelle consistaient en une atrophie de l'utérus, des ovaires et du vagin, observée aux doses représentant environ 0,3 fois la DRH sur la base de l'ASC. Pour ces anomalies, les doses sans effet nocif observé (DSENO) correspondantes représentaient 1,5 fois et 0,1 fois respectivement la DRH sur la base de l'ASC. Chez le rat mâle, une dégénérescence des tubes séminifères et l'absence de libération de spermatozoïdes ont été observées aux doses représentant environ 8 fois la DRH sur la base de l'ASC. Les anomalies chez le singe mâle consistaient en une déplétion en cellules germinales dans les testicules, constatée aux doses représentant environ 0,5 fois la DRH sur la base de l'ASC. Pour ces anomalies, les DSENO correspondantes représentaient 1,4 fois et 0,1 fois respectivement la DRH sur la base de l'ASC. Après une période de récupération de quatre semaines, toutes ces anomalies ont été réversibles, à l'exception des modifications de la muqueuse vaginale chez la rate.
Dans les études de toxicité sur le développement embryonnaire et fœtal, une mortalité embryonnaire et fœtale et une augmentation des pertes post-implantation ont été observées aux doses maternotoxiques. Une fœtotoxicité (poids faible des fœtus, effets sur l'ossification du squelette) et une tératogénicité (anomalies fœtales incluant des œdèmes) ont été constatées aux doses représentant environ 3 fois la DRH sur la base de l'ASC. La DSENO représentait 0,5 fois la DRH sur la base de l'ASC. Le quizartinib est considéré comme potentiellement tératogène.
Études de toxicologie chez l'animal
Dans les études de toxicologie en administration répétée, il a été observé une toxicité sur les organes hématopoïétiques et lymphoïdes consistant en une diminution des taux de cellules sanguines dans le sang périphérique et une hypocellularité médullaire, une toxicité hépatique consistant en une augmentation des transaminases, une nécrose hépatocellulaire et un dépôt de cristaux biréfringents (chez le chien) et une toxicité rénale consistant en une basophilie tubulaire et un dépôt de cristaux biréfringents (chez le rat mâle). Ces anomalies ont été constatées aux doses représentant environ 0,4 fois, 0,4 fois et 9 fois respectivement la DRH sur la base de l'ASC. Les DSENO correspondantes représentaient environ 0,1 fois, 0,1 fois et 1,5 fois respectivement la DRH sur la base de l'ASC.
Les études d'évaluation du risque environnemental ont montré que le quizartinib peut présenter un risque pour le compartiment aquatique.
Études de pharmacologie de sécurité in vitro et chez l'animal
Dans les études de pharmacologie de sécurité cardiovasculaire effectuées chez le singe cynomolgus, le quizartinib a entraîné un allongement de l'intervalle QT aux doses représentant environ 2 fois la DRH de 53 mg par jour sur la base de la Cmax. La DSENO représentait environ 0,4 fois la DRH sur la base de la Cmax. Le quizartinib a inhibé principalement le courant IKs, avec une inhibition maximale de 67,5 % à la concentration de 2,9 µM. L'inhibition maximale du courant IKs par l'AC886 était de 26,9 % à la concentration de 2,9 µM. À la concentration de 3 µM, le quizartinib et l'AC886 ont induit une inhibition statistiquement significative de 16,4 % et 12,0 % respectivement des courants hERG. Ni le quizartinib ni l'AC886 n'ont inhibé les courants INa, INa-L et ICa-L à l'une des concentrations testées.
Ce produit peut présenter un risque pour l'environnement. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription hospitalière.
Prescription réservée aux hématologues ou aux médecins compétents en maladies du sang.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription hospitalière.
Prescription réservée aux hématologues ou aux médecins compétents en maladies du sang.
Comprimé pelliculé (comprimé)
Comprimés pelliculés ronds de couleur jaune, de 10,2 mm de diamètre portant la mention « DSC 512 » gravée en creux sur une face.
Plaquettes prédécoupées unitaires en aluminium/aluminium.
Boîte contenant 56 x 1 comprimé pelliculé.
Chaque comprimé pelliculé contient 26,5 mg de quizartinib (sous forme de dichlorhydrate).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Noyau du comprimé
Hydroxypropylbetadex
Cellulose microcristalline (E 460)
Stéarate de magnésium
Pelliculage
Hypromellose (E 464)
Talc (E 553b)
Triacétine (E 1518)
Dioxyde de titane (E 171)
Oxyde de fer jaune (E 172)